L-丙氨酰-L-氨基丙酸
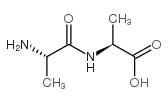
L-丙氨酰-L-氨基丙酸结构式

|
常用名 | L-丙氨酰-L-氨基丙酸 | 英文名 | H-Ala-Ala-OH |
|---|---|---|---|---|
| CAS号 | 1948-31-8 | 分子量 | 160.17100 | |
| 密度 | 1.208 g/cm3 | 沸点 | 402.6ºC at 760 mmHg | |
| 分子式 | C6H12N2O3 | 熔点 | 280-285 °C(lit.) | |
| MSDS | 中文版 美版 | 闪点 | 197.3ºC |
|
Bond dissociation of the dipeptide dialanine and its derivative alanine anhydride induced by low energy electrons.
J. Chem. Phys. 134(5) , 054305, (2011) Dissociative electron attachment to dialanine and alanine anhydride has been studied in the gas phase utilizing a double focusing two sector field mass spectrometer. We show that low-energy electrons (i.e., electrons with kinetic energies from near zero up to... |
|
|
Enhanced susceptibility to antifungal oligopeptides in yeast strains overexpressing ABC multidrug efflux pumps.
Antimicrob. Agents Chemother. 52 , 4057-63, (2008) The susceptibility to several oligopeptide and amino acid antifungals of a Saccharomyces cerevisiae strain carrying multiple deletions in yeast multidrug resistance genes was compared to transformants containing the CDR1, CDR2, or MDR1 genes that encode the m... |
|
|
Infrared spectroscopy of the alanine dipeptide analog in liquid water with DFT-MD. Direct evidence for P(II)/beta conformations.
Phys. Chem. Chem. Phys. 12 , 10198-10209, (2010) Following our previous work [J. Phys. Chem. B. Lett., 2009, 113, 10059], DFT-based molecular dynamics (DFTMD) simulations of 2-Ala peptide (i.e. Ac-Ala-NHMe dialanine peptide analog with methyl group caps at the extremities) immersed in liquid water at room t... |
|
|
Atomic multipoles: electrostatic potential fit, local reference axis systems, and conformational dependence.
J. Comput. Chem. 33(20) , 1673-88, (2012) Currently, all standard force fields for biomolecular simulations use point charges to model intermolecular electrostatic interactions. This is a fast and simple approach but has deficiencies when the electrostatic potential (ESP) is compared to that from ab ... |
|
|
Kinetic mechanism and inhibition of Mycobacterium tuberculosis D-alanine:D-alanine ligase by the antibiotic D-cycloserine.
FEBS J. 280(4) , 1150-66, (2013) D-cycloserine (DCS) is an antibiotic that is currently used in second-line treatment of tuberculosis. DCS is a structural analogue of D-alanine, and targets two enzymes involved in the cytosolic stages of peptidoglycan synthesis: alanine racemase (Alr) and D-... |
|
|
Human PEPT1 pharmacophore distinguishes between dipeptide transport and binding.
J. Med. Chem. 49 , 3636-44, (2006) The human intestinal oligopeptide transporter (PEPT1) facilitates the absorption of dipeptides, tripeptides, and many peptidomimetic drugs. In this study, a large number of peptides were selected to investigate the structural features required for PEPT1 trans... |
|
|
Integrating diffusion maps with umbrella sampling: application to alanine dipeptide.
J. Chem. Phys. 134(13) , 135103, (2011) Nonlinear dimensionality reduction techniques can be applied to molecular simulation trajectories to systematically extract a small number of variables with which to parametrize the important dynamical motions of the system. For molecular systems exhibiting f... |
|
|
Toward canonical ensemble distribution from self-guided Langevin dynamics simulation.
J. Chem. Phys. 134(13) , 134108, (2011) This work derives a quantitative description of the conformational distribution in self-guided Langevin dynamics (SGLD) simulations. SGLD simulations employ guiding forces calculated from local average momentums to enhance low-frequency motion. This enhanceme... |
|
|
A fast parallel clustering algorithm for molecular simulation trajectories.
J. Comput. Chem. 34(2) , 95-104, (2013) We implemented a GPU-powered parallel k-centers algorithm to perform clustering on the conformations of molecular dynamics (MD) simulations. The algorithm is up to two orders of magnitude faster than the CPU implementation. We tested our algorithm on four pro... |
|
|
Umbrella integration with higher-order correction terms.
J. Chem. Phys. 136(23) , 234102, (2012) Umbrella integration is a method to analyze umbrella sampling simulations. It calculates free-energy changes from distributions obtained from molecular dynamics. While it can be formulated on the full sampled distributions, they are generally approximated by ... |