162640-98-4
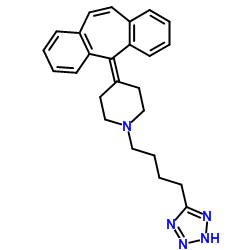
162640-98-4 structure
- Name: AT-56
- Chemical Name: 4-(dibenzo[1,2-a:1',2'-e][7]annulen-11-ylidene)-1-[4-(2H-tetrazol-5-yl)butyl]piperidine
- CAS Number: 162640-98-4
- Molecular Formula: C25H27N5
- Molecular Weight: 397.515
- Catalog: Signaling Pathways Immunology/Inflammation PGE synthase
- Create Date: 2016-08-08 15:03:40
- Modify Date: 2026-06-30 08:52:31
-
AT-56 is a potent, selective and orally active inhibitor of lipocalin-type prostaglandin D synthase (L-PGDS), with an IC50 of 95 μM and Ki of 75 μM. AT-56 could selectively suppress the drowsiness or pain reaction mediated by L-PGDS-catalyzed PGD2[1].
| Name | 4-(dibenzo[1,2-a:1',2'-e][7]annulen-11-ylidene)-1-[4-(2H-tetrazol-5-yl)butyl]piperidine |
|---|---|
| Synonyms |
4-(5H-Dibenzo[a,d][7]annulen-5-ylidene)-1-[4-(1H-tetrazol-5-yl)butyl]piperidine
ICI 215,001 hydrochloride AT-56 |
| Description | AT-56 is a potent, selective and orally active inhibitor of lipocalin-type prostaglandin D synthase (L-PGDS), with an IC50 of 95 μM and Ki of 75 μM. AT-56 could selectively suppress the drowsiness or pain reaction mediated by L-PGDS-catalyzed PGD2[1]. |
|---|---|
| Related Catalog | |
| Target |
IC50: 95 μM (L-PGDS); Ki: 75 μM (L-PGDS)[1] |
| In Vitro | AT-56 (1-30 μM; 10 minutes) dose-dependently inhibits the production of PGD2 in L-PGDS-expressing human medulloblastoma TE-671 cells with an IC50 of about 3 μM[1]. |
| In Vivo | AT-56 ( 1-30 mg/kg; p.o.) suppresses the PGD2 production in the stab-wounded brain[1]. AT-56 (1-10 mg/kg; p.o.) suppresses the L-PGDS-mediated allergic airway inflammation in mice[1]. AT-56 (10 mg/kg; p.o.) exhibits Cmax (2.15 μg/ml), half-life (1.71 h) and high oral bioavailability (82%)[1]. Animal Model: H-PGDS KO mice (14-16weeks, 25-30 g, C57BL/6 strain) with a stab wound brain injury[1] Dosage: 0, 1, 3, 10, 30 mg/kg Administration: P.o. 1 h before the stab wound injury Result: Inhibited the L-PGDS reaction in the brain. Decreased the total amount of PGD2 in the brain to 40% with 30 mg/kg AT-56. Animal Model: Human L-PGDS-overexpressing TG mice (males, 14-16 weeks, 25-30 g)[1] Dosage: 0, 1, 10 mg/kg Administration: P.o. 1 h before and 24 h after the antigen exposure Result: Prevented the eosinophil infiltration by inhibiting transgened human L-PGDS. Animal Model: Male C57BL/6 mice (7 weeks, 22-26 g)[1] Dosage: 10 mg/kg for p.o. and 2 mg/kg for i.v. (Pharmacokinetic Analysis) Administration: P.o. and i.v. administration Result: Oral bioavailability (82%); Cmax (2.15 μg/ml); T1/2 (1.71 h, p.o.); T1/2 (2.35 h, i.v.). |
| References |
| Density | 1.2±0.1 g/cm3 |
|---|---|
| Boiling Point | 620.4±65.0 °C at 760 mmHg |
| Molecular Formula | C25H27N5 |
| Molecular Weight | 397.515 |
| Flash Point | 329.0±34.3 °C |
| Exact Mass | 397.226654 |
| PSA | 57.70000 |
| LogP | 5.84 |
| Vapour Pressure | 0.0±1.8 mmHg at 25°C |
| Index of Refraction | 1.646 |
| Storage condition | -20℃ |
| RIDADR | NONH for all modes of transport |
|---|