| Structure | Name/CAS No. | Articles |
|---|---|---|
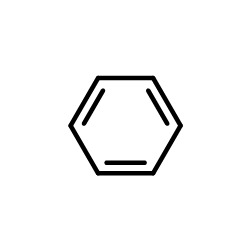 |
benzene
CAS:71-43-2 |
|
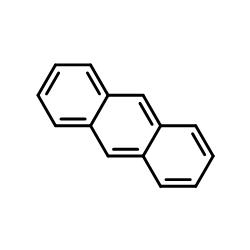 |
Anthracene
CAS:120-12-7 |
|
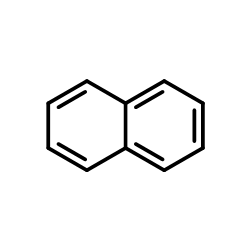 |
Naphthalene
CAS:91-20-3 |
|
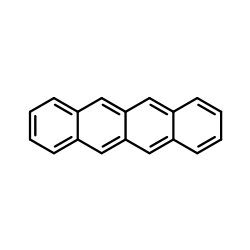 |
Tetracene
CAS:92-24-0 |
|
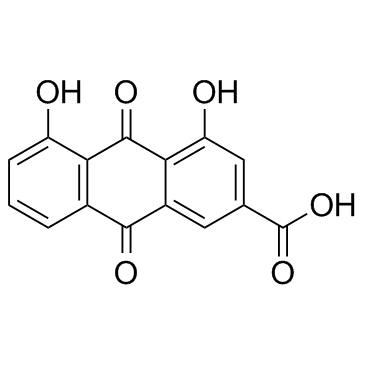 |
Rhein
CAS:478-43-3 |