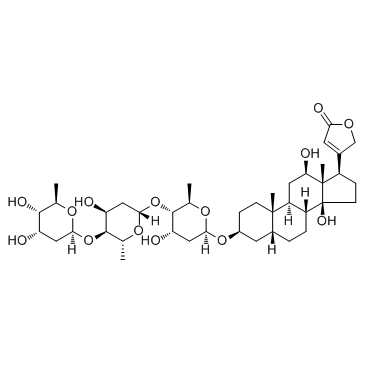
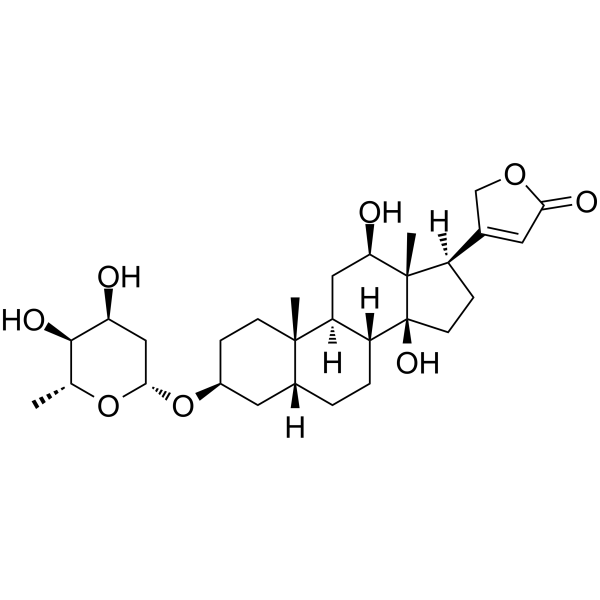
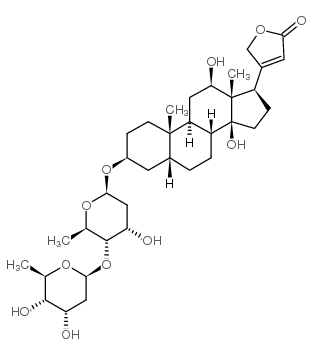
Pyridine-N-oxide occupies an unique position in the quantum mechanical theory of organic reactivity. It undergoes both cationoid and anionoid substitution at the same positions, namely primarily at 4 and to a lesser extent at 2 position. The molecular orbital method of calculating electron density cannot correctly predict the reactivity of this compound, since it cammt predict maximum electrophilic and nucleophilic reactivity for the same position. ...