
- 上海阿拉丁生化科技股份有限公司
- 上海市 上海市 浦东新区
- 产品名:SAR131675
- 纯度:98.0%
- 规格:10mg/1mg/25mg/50mg
- 联系人:阿拉丁

 2881459338
复制
2881459338
复制
- 联系电话:400-620-6333
- 长沙福臻生物科技有限公司
- 湖南省 长沙市 芙蓉区
- 产品名:SAR131675
- 纯度:98.0%
- 规格:100mg/1g
- 联系人:张经理
3756953817
复制

 18229892877
复制
18229892877
复制

- 联系电话:18229892877
- 武汉敬康恩生物医药科技有限公司
- 湖北省 武汉市 东西湖区
- 产品名:SAR131675
- 纯度:98.0%
- 规格:
- 联系人:周经理
1092734927
复制
15871494362
复制

- 联系电话:15871494362
- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:(R)-2-氨基-1-乙基-7-(3-羟基-4-甲氧基-3-甲基丁-1-炔-1基)-N-甲基-4-氧代-1,4-二氢-1,8-萘啶-3-甲酰胺
- 纯度:98.0%
- 规格:
- 联系人:徐经理
351666998
复制
 133 1186 9306
复制
133 1186 9306
复制

- 联系电话:133 1186 9306
查看所有供应商和价格请点击:
1433953-83-3
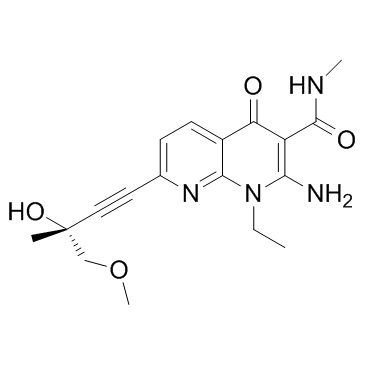
1433953-83-3结构式
| 中文名 | SAR131675 |
|---|---|
| 英文名 | 1,8-Naphthyridine-3-carboxamide, 2-amino-1-ethyl-1,4-dihydro-7-[(3R)-3-hydroxy-4-methoxy-3-methyl-1-butyn-1-yl]-N-methyl-4-oxo |
| 英文别名 |
sar-131675
2-Amino-1-ethyl-7-[(3R)-3-hydroxy-4-methoxy-3-methyl-1-butyn-1-yl]-N-methyl-4-oxo-1,4-dihydro-1,8-naphthyridine-3-carboxamide SAR131675 |
| 描述 | SAR131675是有效,选择性的 VEGFR3 抑制剂,IC50 值为23 nM。 |
|---|---|
| 相关类别 | |
| 靶点 |
VEGFR3:23 nM (IC50) |
| 体外研究 | AR131675对VEGFR-3具有高选择性,与107种受体,酶,离子通道和65种激酶相比。然而,它对VEGFR-2具有中度活性,VEGFR-3/VEGFR-2比率约为10.SAR131675抑制HEFR细胞中VEGFR-3酪氨酸激酶活性和VEGFR-3自身磷酸化,IC50值分别为20和45 nM 。 SAR131675剂量依赖性地抑制由VEGFR-3配体VEGFC和VEGFD诱导的原代人淋巴细胞的增殖,IC50为约20nM。 SSAR131675对一组30个肿瘤和原代细胞没有抗增殖活性,进一步显示其高特异性,表明SAR131675不是细胞毒性或细胞抑制剂[1]。 |
| 体内研究 | SAR131675在小鼠中具有非常好的耐受性,并且在几种原位和同系模型中显示出有效的抗肿瘤作用,包括乳腺4T1癌和RIP1Tag2肿瘤。有趣的是,它显着减少淋巴结侵袭和肺转移,显示其体内抗淋巴管生成活性。 SAR131675显着降低4T1肿瘤中的TAM浸润和聚集[1]。 |
| 激酶实验 | 多孔板预涂有合成聚合物基质poly-Glu-Tyr(polyGT 4:1)。在补充有ATP和二甲基亚砜(DMSO)的激酶缓冲液(10×:50mM HEPES缓冲液,pH7.4,20mM MgCl 2,0.1mM MnCl 2和0.2mM Na 3 VO 4)存在下进行反应,用于阳性对照( C +)或SAR131675(范围为3-1,000 nM)。对于VEGFR-1和VEGFR-3,ATP使用30μM,对于VEGFR-2,ATP使用15μM。用与辣根过氧化物酶偶联的磷酸酪氨酸特异性单克隆抗体(mAb)探测磷酸化的poly-GT,并在黑暗中用HRP显色底物(OPD)显色。然后通过加入100μL1.25mol/ L H 2 SO 4终止反应,并使用Envision分光光度计在492nm处测定吸光度[1]。 |
| 细胞实验 | 将HLMVEC接种在涂有0.3%明胶(每孔5000个细胞)的96孔板中。在不存在或存在SAR131675的情况下,将细胞在RPMI 0.1%FCS中与VEGFA(10ng / mL)VEGFC(300ng / mL),VEGFD(300ng / mL)或FGF2(10ng / mL)一起温育。五天后,用细胞Titer-glo发光细胞活力测定法定量活细胞[1]。 |
| 动物实验 | 小鼠:将用200μgFGF2或PBS浸渍的无菌海绵盘皮下引入麻醉小鼠的背部。前2天将FGF2重新注入海绵中。在海绵植入当天开始每日口服SAR131675(30,100和300mg / kg / d)。七天后,将动物安乐死并取出海绵,收获并在4℃下在RIPA缓冲液中裂解。在6,000×g离心后,收集上清液用于进一步分析[1]。 |
| 参考文献 |
| 密度 | 1.3±0.1 g/cm3 |
|---|---|
| 沸点 | 592.2±50.0 °C at 760 mmHg |
| 分子式 | C18H22N4O4 |
| 分子量 | 358.392 |
| 闪点 | 312.0±30.1 °C |
| 精确质量 | 358.164093 |
| PSA | 119.47000 |
| LogP | -0.61 |
| 外观性状 | 粉末 |
| 蒸汽压 | 0.0±1.8 mmHg at 25°C |
| 折射率 | 1.626 |
| 储存条件 | -20℃ |