Molecular modeling studies of the binding modes of aldose reductase inhibitors at the active site of human aldose reductase.
Y S Lee, Z Chen, P F Kador
文献索引:Bioorg. Med. Chem. 6(10) , 1811-9, (1998)
全文:HTML全文
摘要
Molecular modeling studies using the CHARMM method have been conducted to study the binding modes of aldose reductase inhibitors at the active site of aldose reductase. The energy minimized structures of aldose reductase with six structurally diverse inhibitors (spirofluorene-9,5'-imidazolidine-2',4'-dione (1), 9-fluoreneacetic acid (2), AL1576 (3), 2,7-difluoro-9-fluoreneacetic acid (4), FK366 (5), and Epalrestat (9)) indicate that the side chains of Tyr48, His110, and Trp111 can form numerous hydrogen bonds with either the carboxylate or the hydantoin group of the inhibitors while the side chains of Trp20, Trp111, and Phe122 are positioned to form aromatic-aromatic interactions. Of the three residues (Tyr 48, His 110, and Trp 111) that can form hydrogen bonds with the ionized portion of aldose reductase inhibitors, protonated His110 appears to play an important role in directing charged inhibitors to bind at the active site through charge interaction. Based on the binding mode of the inhibitors and their observed inhibitory activities, pharmacophore requirements for aldose reductase inhibitors are discussed.
相关化合物
| 结构式 | 名称/CAS号 | 分子式 | 全部文献 |
|---|---|---|---|
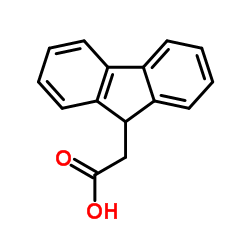 |
9-芴乙酸
CAS:6284-80-6 |
C15H12O2 |
|
Structural basis for the potent calpain inhibitory activity ...
2008-07-24 [J. Med. Chem. 51(14) , 4346-50, (2008)] |
|
Prolonged pheromonotropic activity of pseudopeptide mimics o...
1997-10-31 [Regul. Pept. 72(2-3) , 161-7, (1997)] |
|
Direct determination of adamantanamine in plasma and urine w...
[J. Chromatogr. A. 619(1) , 93-101, (1993)] |
|
Comparison of 9-fluorenylmethoxycarbonyl and 9-fluoreneacety...
1992-08-01 [J. Pharm. Biomed. Anal. 10(8) , 577-86, (1992)] |
|
Automated HPLC analyses of drugs of abuse via direct injecti...
1994-01-01 [Biomed. Chromatogr. 8(2) , 53-62, (1994)] |