Theoretical Studies on the Mechanism and Kinetics of the Reaction of CF3 radical with Oxygen Molecule
Vahid Saheb, Mahdiyeh Javanmardi
Index: 10.1016/j.jfluchem.2018.03.017
Full Text: HTML
Abstract
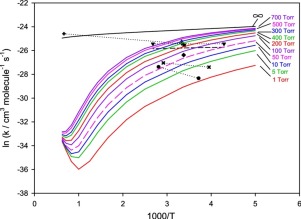
The potential energy surface of the CF3 + O2 reaction is explored by using quantum-chemical combination methods including CBS-QB3, G3B3 and G4. A TST/RRKM model along with a modified strong collision approximation is employed to calculate the thermal rate coefficients for important product channels as a function of temperature and pressure. The calculated results show that the overall rate constant is pressure-dependent over a wide temperature range. At low temperatures, the dominant process is the formation of CF3OO adduct with negative activation energy. However, at higher temperatures, the product channel CF3O + O becomes important. The unimolecular rate coefficients for the thermal decomposition of CF3OO are also computed.