
- 上海化源世纪贸易有限公司
- 上海市 上海市 普陀区
- 产品名:化合物 T28759
- 纯度:99.01%
- 规格:
- 联系人:徐经理

 351666998
复制
351666998
复制


 133 1186 9306
复制
133 1186 9306
复制

- 联系电话:133 1186 9306
查看所有供应商和价格请点击:
116476-13-2
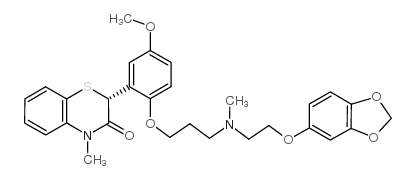
- 常用中文名:司莫地尔
- 常用英文名:semotiadil
- CAS号:116476-13-2
- 分子式:C29H32N2O6S
- 分子量:536.63900
- 相关类别: 原料药 循环系统用药 防治心绞痛药
- 发布时间:2018-08-20 10:24:54
- 更新时间:2025-08-25 08:16:37
-
钙拮抗剂。用于心血管系统疾病的治疗。
| 中文名 | 司莫地尔 |
|---|---|
| 英文名 | (2R)-2-[2-[3-[2-(1,3-benzodioxol-5-yloxy)ethyl-methylamino]propoxy]-5-methoxyphenyl]-4-methyl-1,4-benzothiazin-3-one |
| 中文别名 |
-2-[2-[3-[[2-(1,3-苯并二氧戊环-5-基氧)乙基]甲胺基]丙氧基]-5-甲氧苯基]-4-甲基-2H-1,4-苯并噻嗪-3(4H)-酮
噻莫地尔 |
| 英文别名 |
UNII-DGN08QZ30G
Semotiadilum [INN-Latin] Semotiadil Semotiadil [INN] Semotiadilum LEVOSEMOTIADIL FUMARATE |
| 密度 | 1.263g/cm3 |
|---|---|
| 沸点 | 727.3ºC at 760mmHg |
| 分子式 | C29H32N2O6S |
| 分子量 | 536.63900 |
| 闪点 | 393.7ºC |
| 精确质量 | 536.19800 |
| PSA | 95.00000 |
| LogP | 5.07840 |
| 蒸汽压 | 5.23E-21mmHg at 25°C |
| 折射率 | 1.61 |
| 分子结构 | 1、 摩尔折射率:147.48 2、 摩尔体积(cm3/mol):424.8 3、 等张比容(90.2K):1134.0 4、 表面张力(dyne/cm):50.7 5、 极化率(10-24cm3):58.46 |
| 计算化学 | 1.疏水参数计算参考值(XlogP):5.1 2.氢键供体数量:0 3.氢键受体数量:8 4.可旋转化学键数量:11 5.互变异构体数量:2 6.拓扑分子极性表面积95 7.重原子数量:38 8.表面电荷:0 9.复杂度:761 10.同位素原子数量:0 11.确定原子立构中心数量:1 12.不确定原子立构中心数量:0 13.确定化学键立构中心数量:0 14.不确定化学键立构中心数量:0 15.共价键单元数量:1 |
方法1:从消旋的化合物(I)出发,合成出最终的消旋产物后,再进行拆分。化合物(I)(30.1g,0.10mo1)溶于60ml干燥二甲基甲酰胺,在0℃滴加氢化钠(60%的矿物油悬浮物,4.80g,0.12mo1)在40ml干燥二甲基甲酰胺的溶液,然后在室温下搅拌30min。加入1-溴-3-氯丙烷 (18.9g,0.12mo1)在30ml干燥二甲基甲酰胺中的溶液,在室温下搅拌2h。倾入700ml水中,用乙酸乙酯提取,提取液用水和盐酸洗,无水硫酸镁干燥,然后减压浓缩。沉淀出的结晶用甲醇洗,得33.3g化合物(Ⅱ),收率88%,熔点97-99℃。
化合物(Ⅱ)(11.3g,30.0mo1) N-甲基-N-[2-[3,4-(亚甲基二氧)苯氧基]乙基]胺(Ⅲ,6.15g,31.5mmo1)溶于60ml干燥二甲基甲酰胺,加入碳酸氢钠(7.56g,90mmo1)和碘化钠(8.99g,60mmo1),在70~80℃搅拌3h。冷却后,加入200ml水处理,再用乙酸乙酯提取。提取液用水和盐水洗,无水硫酸镁干燥,真空浓缩。剩余的油状物用硅胶层析,氯仿-甲醇(50:1)洗脱,得到油状的消旋的司莫地尔产物。将其溶于乙酸乙酯,加入稍过量的富马酸的乙醇溶液。过滤收集沉淀出的结晶,用乙醇重结晶,可得13.3g消旋的富马酸司莫地尔,收率68%。
消旋的司莫地尔(10.73g,20.0mmo1)溶于15ml丙酮,加入(+)-扁桃酸(1.52g,10.0mmo1)在18ml环己烷的溶液,在室温下搅拌1天。过滤收集析出的沉淀,干燥,得5.92g白色粉末的(+)-富马酸盐;再用丙酮重结晶,得4.40g白色结晶,熔点114~115℃,[α]D23+206°(C=1.1,二甲亚砜)。滤液减压浓缩,剩余的油状物溶于40ml二氯甲烷,加入20ml 0.5mol/L氢氧化钠处理,然后用盐水洗,无水硫酸镁干燥。过滤,浓缩,剩余物溶于14ml丙酮。加入(-)-扁桃酸(1.52g,10.0mmo1)在14ml环己烷的溶液,室温搅拌1h,再在冰箱中放置2h。过滤收集析出的沉淀,得5.97g白色粉末的(-)-扁桃酸盐;再用丙酮重结晶,得4.67g白色结晶,熔点114~115℃,[α]D23-214°(C=1.0,二甲亚砜)。
前面得到的(+)-扁桃酸盐(4.40g,6.38mmo1)悬浮于27ml二氯甲烷,加入饱和碳酸氢钠处理后,再用盐水洗,无水硫酸镁干燥。过滤,浓缩,残物溶于9ml乙酸乙酯,加入富马酸(0.74g,6.38mmo1)在18ml乙醇的热溶液,室温搅拌1h,再冰箱中放置10h。过滤收集沉淀,用18ml乙醇重结晶,得3.38g(+)-司莫地尔(100%ee),收率26%(以消旋司莫地尔计)。同样,从(-)-扁桃酸盐(4.67g,6.78mmo1)得到3.59g(-)-司莫地尔(100%ee),收率28%(以消旋司莫地尔计)。
方法2:化合物(I)(202.7g,673mmo1)和化合物(Ⅳ)(312.3g,1.01mo1)溶于 850ml二甲基甲酰胺,在冰浴冷却下,加入N,N’-二环己基碳化二亚胺(DCC,208.3g,1.01mo1)在500ml吡啶的溶液和4-(二甲氨基)吡啶(13.3g,1.01mo1),在室温搅拌4h。将反应液倾入水-乙酸乙酯(6 L:1.7 L)的混合液中,加入草酸(30.3g,337mmo1),并在室温下搅拌15min。过滤除去沉淀,滤液分层,水层用乙酸乙酯提取。提取液和有机层合并,用水及盐水洗,无水硫酸镁干燥,浓缩,得到棕色油状物。用硅胶层析,己烷-苯-乙酸乙酯(1:5:2)洗脱,得171.6g化合物(V),收率48%;和179.6g化合物(Ⅵ),收率45%。
在0℃,将硼氢化钠(58.6g,1.55mo1)加到二氯化钙(179.8g,1.62mo1)在460ml四氢呋喃的悬浮液中,得到硼氢化钙溶液。在-10℃,加入化合物(V)(183.1g,309mmol)在1.2L乙醇的溶液和氯化铵(165.8g,3.10mo1),再搅拌12h。用2.5L 1mol/L盐酸处理后,减压浓缩,再用乙酸乙酯提取。提取液用盐水洗,无水硫酸镁干燥,减压浓缩。剩余物硅胶柱层析,用己烷-乙酸乙酯-氯仿(1:1:8)洗脱,用苯重结晶,得69.9g(+)-(I),收率75%,熔点161~162℃,[α]D23+40°(C=1.17,氯仿)。
(+)-(Ⅰ)(10.0g,33.2mmo1)和偶氮二羧酸二乙酯(17.4g,99.6mmo1)溶于干燥二甲氧基乙烷-四氢呋喃(10ml:1.55m1)混合液。在氮气保护、0~5℃和搅拌下,于25min中将上述混合液滴加到3-溴-1-丙醇(13.8g,99.6mmo1)和三苯膦(26.1g,99.6mmo1)在100ml干燥二甲基甲酰胺的溶液中。用30ml0.5mol/L盐酸处理后,再减压浓缩。过滤除去析出的结晶,滤液用硅胶层析,己烷-苯-乙酸乙酯(5:15:1)洗脱。再用异丙醇重结晶,得2.53g(+)-(Ⅶ),收率18.1%。
(+)-(Ⅶ)(2.43g,5.75mmo1)溶于60ml丙酮,加入碘化钠(8.62g,57.5mmo1),回流20h。蒸出溶剂,剩余物溶于乙酸乙酯,用水及盐水洗,无水硫酸镁干燥。蒸出溶剂,剩余物溶于25ml干燥二甲基甲酰胺,加入化合物(Ⅲ)(1.46g,7.48mmo1)和三乙胺(0.58g,5.75mmo1),在50℃搅拌1.5h。倾入水中,用乙酸乙酯提取。提取液用盐水洗,无水硫酸镁干燥,蒸出溶剂。残液用硅胶柱层析,用氯仿-甲醇(50:1)洗脱,得到油状的(+)-司莫地尔。将其溶于乙酸乙酯,加入稍过量的富马酸的乙醇溶液,过滤析出的结晶,再用乙醇重结晶,得2.1lg(+)-富马酸司莫地尔,收率56.2%。
同样可从(-)-(I)得到(-)-富马酸司莫地尔。