A computational study on DNA bases interactions with dinuclear tetraacetato-diaqua-dirhodium(II,II) complex.
Jaroslav V Burda, Jiande Gu
文献索引:J. Inorg. Biochem. 102(1) , 53-62, (2008)
全文:HTML全文
摘要
In our study, we have determined the thermodynamic behavior for the replacement reaction of one and two acetyl-ligands from the diaqua-tetrakis(mu-acetylato)dirhodium(II,II) complex by purine DNA bases. The complexes were optimized at the density functional theory (DFT) level with the B3LYP functional. Stuttgart-Dresden pseudopotentials were used for the description of the Rh atoms. Most of the replacement reactions are mildly exothermic, delta G is up to 12 kcal/mol for the first acetyl-ligand and up to 8 kcal/mol for the second ligand replacement. For all explored complexes, stabilization and bonding energies were computed together with selected electronic properties. Adenine base coordinates to the dirhodium complex slightly more firmly than guanine. In head-to-tail conformation the two guanines are better stabilized (by about 8 kcal/mol) than in head-to-head arrangement due to minimization of sterical repulsion of both bases. We have shown that the bonding energy of axial water ligands is very small (up to 13 kcal/mol), resembling more H-bonds than dative coordination. Despite the larger stabilization energies of adenine-containing complexes, the thermodynamic parameters of the studied replacement reactions are more favorable in case of guanine complexes. Higher exothermicity is connected with easier deprotonization of guanine N1-site in comparison with N6-site of adenine in accord with experimental data.
相关化合物
| 结构式 | 名称/CAS号 | 分子式 | 全部文献 |
|---|---|---|---|
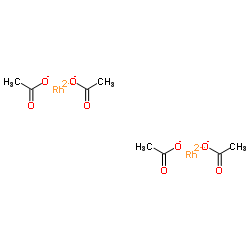 |
醋酸铑(II)二聚体
CAS:15956-28-2 |
C8H12O8Rh2 |
|
Rhodium acetate/base-catalyzed N-silylation of indole deriva...
2012-09-25 [Chem. Commun. (Camb.) 48(74) , 9269-71, (2012)] |
|
One-pot protocol to functionalized benzopyrrolizidine cataly...
2015-01-02 [Org. Lett. 17(1) , 66-9, (2015)] |
|
Unprecedented head-to-head conformers of d(GpG) bound to the...
2003-09-03 [J. Am. Chem. Soc. 125(35) , 10703-13, (2003)] |
|
Distal renal tubular acidosis associated with anion exchange...
2007-06-01 [Am. J. Kidney Dis. 49(6) , 841-850.e1, (2007)] |
|
Interaction of tetra-mu-acetatodirhodium(II) with human seru...
1995-04-01 [J. Inorg. Biochem. 58(1) , 69-77, (1995)] |