| 结构式 | 名称/CAS号 | 全部文献 |
|---|---|---|
 |
(|S|)-(+)-α-苯甘氨酸
CAS:2935-35-5 |
|
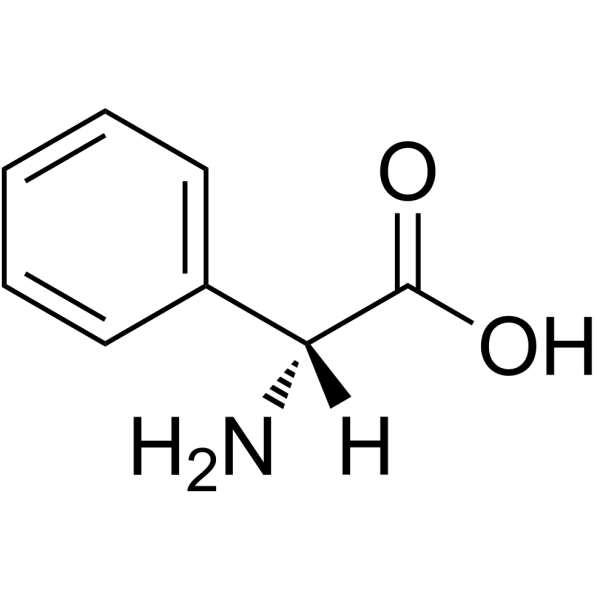 |
D-(-)-α-苯基甘氨酸
CAS:875-74-1 |
|
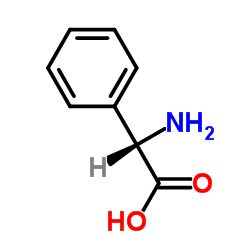 |
DL-苯甘氨酸
CAS:2835-06-5 |