Conformational analysis of bis(methylthio)methane and diethyl sulfide molecules in the liquid phase: reverse Monte Carlo studies using classical interatomic potential functions.
Orsolya Gereben, László Pusztai
文献索引:J. Phys. Condens. Matter 25(45) , 454201, (2013)
全文:HTML全文
摘要
Series of flexible molecule reverse Monte Carlo calculations, using bonding and non-bonding interatomic potential functions (FMP-RMC), were performed starting from previous molecular dynamics results that had applied the OPLS-AA and EncadS force fields. During RMC modeling, the experimental x-ray total scattering structure factor was approached. The discrepancy between experimental and calculated structure factors, in comparison with the molecular dynamics results, decreased substantially in each case. The room temperature liquid structure of bis(methylthio)methane is excellently described by the FMP-RMC simulation that applied the EncadS force field parameters. The main conformer was found to be AG with 55.2%, followed by 37.2% of G(+)G(+) (G(-)G(-)) and 7.6% of AA; the stability of the G(+)G(+) (G(-)G(-)) conformer is most probably caused by the anomer effect. The liquid structure of diethyl sulfide can be best described by applying the OPLS-AA force field parameters during FMP-RMC simulation, although in this case the force field parameters were found to be not fully compatible with experimental data. Here, the two main conformers are AG (50.6%) and the AA (40%). In addition to findings on the actual real systems, a fairly detailed comparison between traditional and FMP-RMC methodology is provided.
相关化合物
| 结构式 | 名称/CAS号 | 分子式 | 全部文献 |
|---|---|---|---|
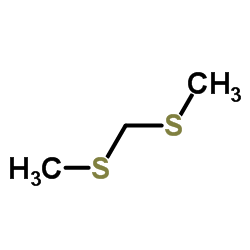 |
二甲硫基甲烷
CAS:1618-26-4 |
C3H8S2 |
|
Olfactometry Profiles and Quantitation of Volatile Sulfur Co...
2015-09-02 [J. Agric. Food Chem. 63 , 7511-21, (2015)] |
|
One-dimensional coordination polymers incorporating silver(I...
2007-04-16 [Inorg. Chem. 46(8) , 3185-91, (2007)] |
|
Truffle thio-flavours reversibly inhibit truffle tyrosinase.
2003-03-14 [FEMS Microbiol. Lett. 220(1) , 81-8, (2003)] |