| 结构式 | 名称/CAS号 | 全部文献 |
|---|---|---|
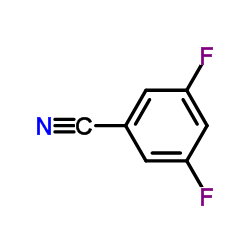 |
3,5-二氟苯甲腈
CAS:64248-63-1 |
| 结构式 | 名称/CAS号 | 全部文献 |
|---|---|---|
|
|
3,5-二氟苯甲腈
CAS:64248-63-1 |