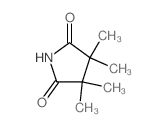
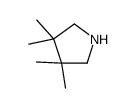
Abstract Molecular modeling by molecular mechanics (MM2) calculations were used to predict conformational preferences of cyclic N-nitrosamines. The results were compared with 1 H NMR data. The observed geminal coupling constants 2 J were considerably stronger for syn than for anti α-methylene protons and thus appear to be very useful for configurational predictions. The long-range 4 J couplings across the nitrogen atom were observed directly ...