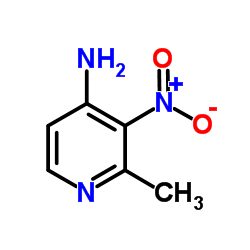
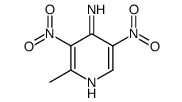
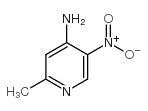
Abstract The synthesis of the new C-nucleoside 6-deazaformycin A was achieved through the condensation of a suitably substituted lithiated 2-picoline with 2, 3, 5-tri-O-benzyl-d- ribonolactone, borohydride reduction of the resulting hemiacetals, followed by intramolecular Mitsunobu cyclization of the carbinols, manipulation of the protecting groups, and subsequent ring closure to result in the formation of 7-amino-3-(β-d-ribofuranosyl) ...