Shedding Light on Important Waters for Drug Design: Simulations versus Grid-Based Methods
Denis Bucher, Pieter Stouten, Nicolas Triballeau
文献索引:10.1021/acs.jcim.7b00642
全文:HTML全文
摘要
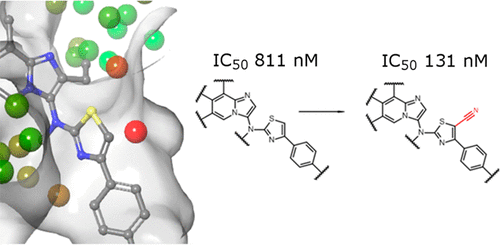
Water molecules play an important role in the association of drugs with their pharmaceutical targets. For this reason, calculating the energetic contribution of water is essential to make accurate predictions of compounds’ affinity and selectivity. Water molecules can also modify the binding mode of compounds by forming water bridges, or clusters, that stabilize a particular orientation of the ligand. Several computational methods have been developed for solvent mapping, but few studies have attempted to compare them in a drug design context. In this paper, four commercially available solvent mapping tools (SZMAP, WaterFLAP, 3D-RISM, and WaterMap) are evaluated on three different protein targets. The methods were compared by looking at their ability to predict the structure–activity relations of lead compounds. All methods were found to be useful to some degree and to improve the predictions from docking alone. However, the only simulation-based approach tested, WaterMap, was found in some cases to be more accurate than grid-based methods.