| Structure | Name/CAS No. | Articles |
|---|---|---|
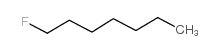 |
1-fluoroheptane
CAS:661-11-0 |
| Structure | Name/CAS No. | Articles |
|---|---|---|
|
|
1-fluoroheptane
CAS:661-11-0 |