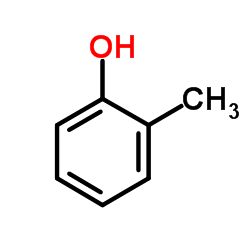
An ab initio molecular orbital theory has been applied to the elucidation of the hydrogen abstraction mechanism of phenolic antioxidants in the chain process of autoxidations. The optimum structures of o-, m-, and p-substituted phenols, of peroxides, and of those compounds in the transition states were obtained with a Hartree-Fock/STO-3G basis set. From the values of the enthalpy (ΔH), activation (E a), and OH bond dissociation (D (O–H)) ...