Theoretical Studies of the Formation Mechanisms, Thermodynamic Stabilities, and Water-Exchange Reactivities of Aluminum-Salicylate Complexes in Aqueous Solution
Shaonan Dong, Wenjing Shi, Jing Zhang, Shuping Bi
Index: 10.1021/acsearthspacechem.7b00141
Full Text: HTML
Abstract
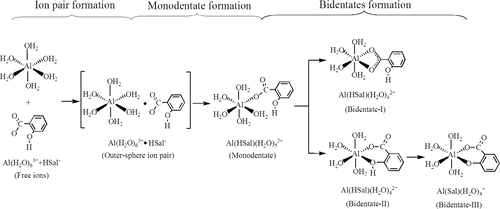
The formation mechanisms, thermodynamic stabilities, and water-exchange reactivities of 1:1 monomer aluminum–salicylate (Al–salicylate) complexes in acidic aqueous solution are investigated using the density functional theory-quantum chemical cluster model (DFT-CM) method. (1) The formation pathways for possible monodentate and bidentate Al–salicylate configurations are modeled with the gas phase-supermolecule-polarizable continuum model (GP-SM-PCM). It shows that the formation pathways for the Al–salicylate complexes follow the Eigen-Wilkins mechanism, where the dissociation of an inner-shell coordinated water of Al3+ is the rate-determining step. (2) The formation constants Kaq for different Al–salicylate configurations are estimated based on the total Gibbs free energy changes ΔG° for their overall formation pathways. It is indicated that in the acidic aqueous solution at pH ∼ 3, the main existence form of the 1:1 monomer Al–salicylate complex is the phenol-deprotonated bidentate Al(Sal)(H2O)4+ with six-membered ring. Its log Kaq is calculated as 13.8, in good agreement with the literature values of 12.9–14.5. (3) The water-exchange reactions are modeled for different Al–salicylate configurations. The water-exchange rate constant for Al(Sal)(H2O)4+ is estimated as log kH2O = 3.9 s–1, close to the experimental value of 3.7 s–1. It proves again that this configuration is the dominant form under experimental conditions.